I) Method Development
F) Conformational and Structural Analysis
- Ring Puckering Coordinates
- Development of Coordinates for special internal processes: Bond-pseudorotation
2) Development of Coordinates for special internal processes: Bond-pseudorotation
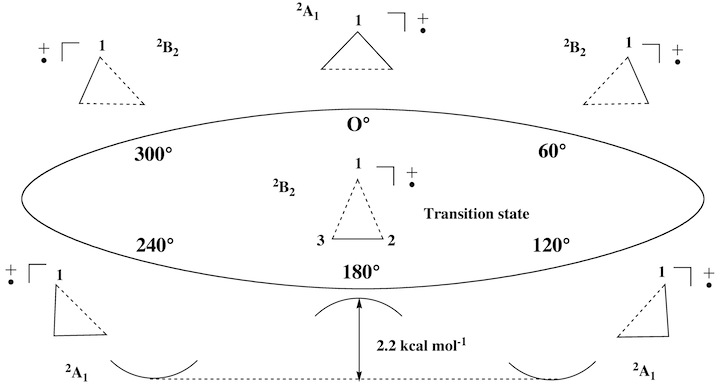
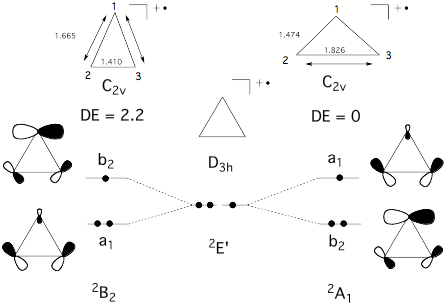
Conformations and conformational changes of puckered rings are mostly described in terms of dihedral angles and dihedral angle changes, although a mathematically unique and consistent way is only provided by the ring puckering coordinates defined by Cremer and Pople (in the literature called CP puckering coordinates). The basic principle of the CP coordinates is to define basis conformations and to represent the given conformation of a puckered monocyclic ring as a linear combination of basis conformations. Ring inversion, semi-inversions, and pseudorotational modes are easy to describe in this way. Our work on the rearrangement modes of the barbaralyl cation led to the question whether rearrangements of planar monocyclic (2 dimensions) or polycyclic molecules (3 dimensions) can be described in a similar way. While this seems to be first just a mathematical question, its chemical relevance becomes clear when considering that with an unique mathematical definition a better understanding of already known processes and the discovery of so far unknown chemical (conformational and configurational) processes is possible. For example, it should be possible that under certain circumstances a bond (or even several bonds) can pseudorotate through a planar ring. We interpret the ESR signals of cyclopropyl radical cations in this way and we predict that bond-pseudorotation should generally exist in Jahn-Teller unstable, planar ring molecules. We carried out the search for bond-pseudorotating molecules in three steps: a) setting up appropriate mathematical formulas for describing bond pseudorotation in 2 and 3 dimensions; b) search and investigation of planar rings with bond pseudorotation; c) work on a general description of configurational and conformational processes including bond-pseudorotation, ring pseudorotation, Berry pseudorotation, wheel-driven pseudorotation, etc.
G) Analysis of the Potential Energy Surface
Finding the Transition State of Quasi-barrierless Reactions by a Growing String Method for Newton Trajectories
A method for finding a transition state (TS) between a reactant minimum and a quasi-flat, high dissociation plateau on the potential energy surface is developed. The method is based on the search of a growing string (GS) along reaction pathways defined by different Newton trajectories (NT). Searches with the GS-NT method always make it possible to identify the TS region because monotonically increasing NTs cross at the TS or, if not monotonically increasing, possess turning points that are located in the TS region. The GS-NT method can be applied to quasi-barrierless and truly barrierless chemical reactions. Examples are the dissociation of methylenecyclopropene to acetylene and vinylidene, for which a small barrier far out in the exit channel is found, and the cycloaddition of singlet methylene and ethene, which is barrierless for a broad reaction channel with Cs-symmetry reminding of a mountain cirque formed by a glacier.
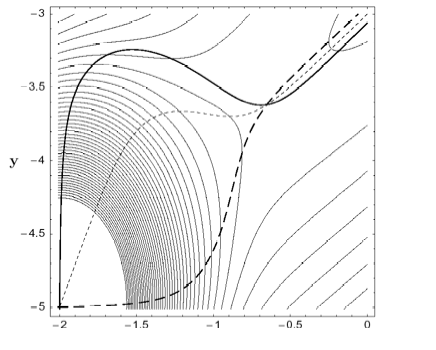

Figure 1. Two-dimensional (2D) model PES a) with a small energy barrier (above) and b) without a barrier in the flat dissociation channel (below). Three different Newton trajectories are included (bold solid, bold dashed, dashed lines), which either cross at the TS (above) or concentrate in the dissociation channel (below).