I) Method Development
B) Density Functional Theory
- SIC-DFT: Kohn Sham DFT with a Self-interaction Correction
- CASDFT - The simultaneous treatment of non-dynamical and dynamical correlation effects
1. SIC-DFT: Kohn Sham DFT with a Self-interaction Correction
Nowadays, DFT is one of the most widely used methods in quantum chemistry for reasons, which have been described in many review articles. Our research group has for several years a strong DFT research program, which covers among others the development of new DFT methods for the calculation of magnetic properties (NMR chemical shifts with DFT-IGLO, NMR spin-spin coupling constants with CPDFT, magnetic susceptibilities and susceptibility exaltations), the description of van der Waals complexes with new DFT functionals, the calculation of singlet biradicals with the ROSS method, the calculation of multi-reference systems with CAS-DFT, the investigation of UDFT as opposed to UHF with the on-top pair density, the evaluation of S2 within DFT, etc. Apart from this, many applications with DFT have been carried out in our research group so that we have a detailed overview over the advantages and disadvantages of DFT. As for the latter, it is a drastic failure of DFT not to be able to correctly describe the simplest diatomic molecule, namely H2+ because DFT suffers from the well-known self-interaction error (SIE: Coulomb and exchange energy of the one-electron self-interaction do not cancel out).
The SI problem is well-described in the literature (see, e.g. J.A. Pople et al., Chem. Phys. Lett., 221, 100, 1994), however, there is still no general description of all its consequences and a self-consistent SI corrected (SIC) DFT method although suggested by Perdew and Zunger (Phys. Rev. B, 23, 5048, 1981) is not generally available.
Our SIC project comprises five goals: 1) Description of the consequences of the SI error for dissociation and other reaction energies, charge distributions, geometries, NMR chemical shift and NMR coupling constants. - 2) Programming of a self-consistent SIC-DFT method: One has to consider that SIC-DFT (contrary to normal DFT) is based on an orbital (density)-dependent Fock operator so that the SCF iterations become more difficult. The minimum conditions can be set up in such a way that they can be solved either by iterated diagonalization or by a direct minimization using steepest descent or conjugate gradient approaches. The first method is faster but has the disadvantage to lead to delocalized orbitals while SIC-DFT requires in some cases localized orbitals. The second method is less efficient but converges more safely and can be equally applied to localized and delocalized MOs, which is the reason why will focus on it. - 3) Development and testing of new XC functionals, which optimally perform in connection with SIC-DFT. - 4) Application of SIC-DFT in connection with mechanistic studies described for projects 1-3. - 5) Testing of the performance of SIC-DFT in the case of van der Waals complexes.
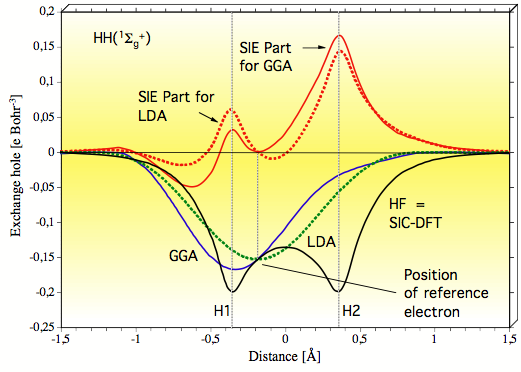
Figure 1. Graphical representation of the exchange hole calculated for H2 in its ground state along the bond axis at the HF, SIC-DFT (black line), LDA (dashed green line), and GGA levels of theory (blue line) for a position of the reference electron as indicated. The SIE part of the LDA exchange hole is given by red the lines in normal print. All calculations with a cc-pVTZ basis set at the experimental geometry.
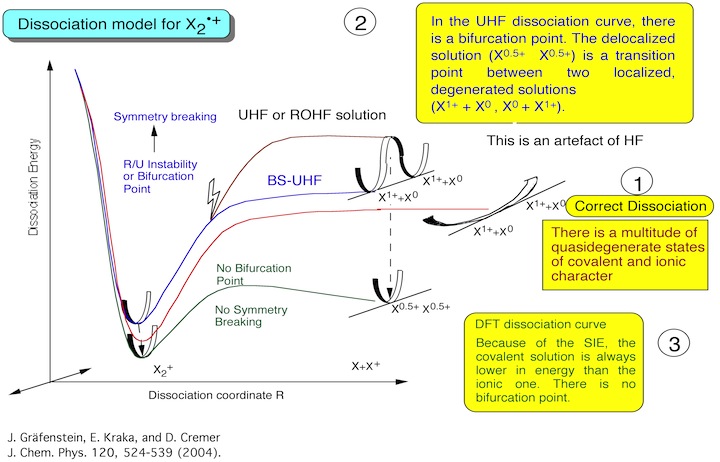
2. CASDFT - The simultaneous treatment of non-dynamical and dynamical correlation effects
The goal of our work is to develop a calculational scheme that provides a balanced description of non-dynamic and dynamic electron correlation by combining a multi-reference (CAS: complete active space) wave-function approach with a density functional theory (DFT) correlation functional for the description of the dynamic correlation. The central problem with such a CAS-DFT approach is to avoid a double-counting of dynamic correlation effects. We started from the approach suggested by Miehlich, Savin, and Stoll (MSS). MSS developed a probe that allows to locally assess the size of the active space (i.e. the number and effectiveness of the extra orbitals provided to describe correlation effects), and hence the amount of dynamical correlation contained in the multi-reference wave function so that one can reduce the DFT correlation energy by local scaling. The approach is based on an appropriate generalization of the Local-Density approximation (LDA).
We extended the probe suggested by MSS in three ways: (i) The difference between active and inactive orbitals in the CAS wave-function is accounted for (MSS assume implicitly that all occupied orbitals are in the active space). (ii) Closed-shell and open-shell systems are treated in a balanced way. Also in distinction to MSS we describe the DFT correlation energy with the Colle-Salvetti functional, which is the most natural choice when a multi-reference wave function is available. (iii) The suggestion of Stoll, Pavlidou and Preuss (SPP) to improve the accuracy of DFT correlation energies was applied to the definition of the probe function. Test calculations for model systems (e.g. methylene) confirmed that our approach improves the balance between CAS-DFT descriptions of different states. However, there remains an inconsistency between the description of high-spin and low-spin open-shell states. This can be remedied by another change: (iv) Instead of the real spin-resolved electron density, the unpaired-electron density by Davidson and Staroverov is used to detect open-shell states.
CAS-DFT (Complete Active Space Density Functional Theory) is presented as a method that allows an economical, simultaneous treatment of non-dynamical and dynamical correlation effects for electronic systems with multi-reference character. Central problems of CAS-DFT concern the effective coupling between wave function and DFT method, the double counting of dynamical correlation effects, the choice of the proper input quantities for the DFT functional, the balanced treatment of core and active orbital correlation, of equal-spin and opposite-spin correlation effects, and the inclusion of spin polarization to handle closed- and open-shell systems in a balanced way. We present CAS-DFT2(CS,SPP,FOS,DS) (CAS-DFT using level 2 for the distinction of core and active orbital correlations, carried out with the Colle-Salvetti functional, using the Stoll-Pavlidou-Preuss functional for equal-spin correlation corrections, including spin-polarization in the scaling procedure, and correcting with the Davidson-Staroverov density for low-spin cases). The method is free of any self-interaction error and size-extensive provided the active space is properly chosen. For the three lowest states of methylene, stringent and less stringent tests are used to demonstrate the performance of the new CAS-DFT method for six different active spaces.
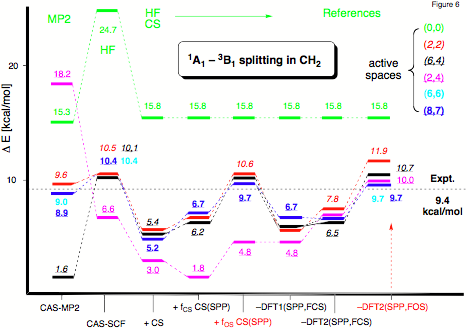
Figure 6. Changes in the excitation energy E(1A1) - E(3B1) of CH2 for an increasing active space calculated at different levels of theory with a 6-311++G(2df,2pd) basis set. CS: Colle-Salvetti functional; FCS and FOS: scaling factor f calculated for a closed-shell and open-shell situation, respectively; SPP: Stoll-Pavlidou-Preuss functional for elimination of equal-spin correlation; levels 1 and 2: different levels of distinguishing between core and active electron correlation.
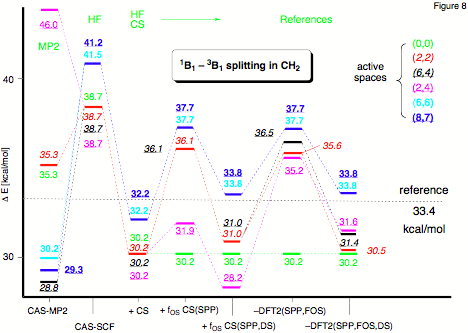
Figure 8.Changes in the excitation energy E(1A1) - E(3B1) of CH2 for an increasing active space calculated at different levels of theory with a 6-311++G(2df,2pd) basis set. CS: Colle-Salvetti functional; FCS and FOS: scaling factor f calculated for a closed-shell and open-shell situation, respectively; SPP: Stoll-Pavlidou-Preuss functional for elimination of equal-spin correlation; DS: Davidson-Staroverov correction for low-spin cases;levels 1 and 2: different levels of distinguishing between core and active electron correlation.