X) Exact Geometries
Determination of Exact Geometries from Quantum Chemical Investigations
The knowledge of molecular equilibrium geometries is a basic requirement for the understanding of the electronic structure, thermodynamic stability, and chemical reactivity of molecules. In the last century, experimental work has led to an impressive collection of reliable geometrical data of molecules utilizing spectroscopic (microwave, infrared, Raman, etc.) and diffraction methods (electron diffraction, X-ray diffraction, neutron diffraction, etc.). These techniques yield information on average nuclear positions in ground and excited vibrational states of a molecule, thermally averaged values of distances and angles, or molecular parameters such as effective ground state rotational constants that are related to the geometrical parameters of a molecule. Because of the Heisenberg principle, it is not possible to measure equilibrium geometries re directly. These have to be derived from experimentally based molecular structures (ro, rg, ra, rz, also rs, rm, ra, rv) using additional information such as the molecular force field, rotation-vibration couplings, coriolis couplings, etc. The equilibrium geometry re of a molecule, although not measurable, is well-defined and directly comparable to calculated geometries based on the use of reliable quantum chemical methods. Actually, calculated re geometries can be used to predict, verify or improve measured geometries in two different ways.
- Experimentally derived re geometries can be used to test the reliability of a given quantum chemical method. If the latter is found to be reliable it can be applied to determine the equilibrium geometry of molecules, for which it is not possible to obtain an experimentally based re geometry.
- Quantum chemical methods can be used to calculate the rz, ro, rg, etc. geometry of a molecule, which can be directly compared with measured geometries. If the method in question is known to lead to reliable geometries (point 1) this approach is the basis for directly checking, complementing or predicting measured geometries.
In recent work, we focused (in collaboration with J. Gauss and J. Stanton) on task 2) to support the work of spectroscopists and diffractionists in structure determination. A typical examples is the structure determination of cyclopropane [1], for which an exact experimental re geometry could not be obtained.
In parallel work, we tried to get a better understanding of quantum chemically obtained geometries. Whereas experimental geometries depend on the measuring technique, the environment of the molecule (gas phase, liquid, solution or solid), and the state of the molecule (electronic, vibrational, rotational), calculated geometries of a given molecular state depend on the quantum chemical method applied, in particular the amount of electron correlation accounted for, and (within any LCAO approach) on the number of basis functions employed. Since electron correlation effects and basis set effects both influence the calculated geometry, it is difficult to separate both effects. Forty years of quantum chemical work have led to considerable knowledge about the accuracy of molecular properties calculated with a given basis set and a given method. Nevertheless, it is still an art in itself to choose method and basis set for the calculation of a molecular geometry in such a way that the best compromise between computational cost and accuracy of geometrical parameters is obtained.
In a recent project, we solved the method/basis set problem in a somewhat different way from what is usually done, namely by eliminating one of the two factors determining the accuracy of a calculated molecular geometry. This was accomplished by determining that molecular geometry, which is obtained when the basis set used with a particular quantum chemical method becomes infinitely large, i.e. when the complete basis set (CBS) limit is reached. Geometries obtained at the method/CBS level do no longer depend on deficiencies of a truncated basis set and, therefore, reflect directly the accuracy of the method chosen, which in turn is related to the electron correlation effects covered by it.
Although the advantages of using CBS limit geometries in quantum chemical investigations is obvious, it is not obvious how to determine them. The computational cost of single determinant methods, which cover just pair correlation effects (as described by double (D) excitations) scale with O(M5)or O(M6) where M denotes the number of basis functions. The inclusion of three-electron correlation effects (described by triple (T) excitations) leads to O(M7) and O((M8) methods whereas the inclusion of connected four-electron correlations (described by quadruple (Q) excitations) already requires O(M9) and O(M10) methods. Utilizing supercomputers, a pair-correlation method can still be carried out with more than 1000 basis functions, the limit for the simplest T excitation methods is at about 250 basis functions (but an extension to 450 basis functions is possible with direct methods), and reduces to just 50-60 basis functions in the case of the Q excitation methods.
Since a hextuple zeta basis augmented with d-, f-, g-, h-, and i-type polarization functions such as [7s6p5d4f3g2h1i/6s5p4d3f2g1h] can be considered to lead to energies and geometries close or equal to the basis set limit of a given ab initio method, a geometry optimization of a small molecule such as methane would include already 504 basis functions, which is too large even for the simplest T method. Clearly, CBS limit geometries have to be determined in another way than just extending M to large enough values so that the CBS limit of a given property is reached within calculational accuracy. However, one can make use of the fact that calculated geometrical parameters Q monotonically converge with increasing basis set to the CBS value Q(CBS) = Q(M = ∞) and that this convergence behavior can be modeled with appropriate exponential functions. Work by Cremer and co-workers has confirmed this in several cases and the procedures worked out by members of the PCTC constitute the basis for the present investigation.
We calculated CBS limit geometries as obtained with different correlation corrected methods. The goal of this work was to determine the method that leads to the most accurate re-geometries. For this purpose, we selected a set of suitable test molecules for which accurate experimentally based equilibrium geometries are known. Since all molecules considered represent closed/open shell systems without significant multi-reference character, we applied single determinant theory starting from a Hartree-Fock (HF) reference wave function. The correlation corrected methods used were limited to single determinant many body perturbation theory (MBPT) and single determinant coupled cluster theory (CC) because these theories lead to hierarchies of size-extensive methods, which make it possible to investigate the influence of electron correlation effects of increasing order on the accuracy of calculated geometrical parameters. Such a relationship cannot be deduced from correlation corrected methods based on density functional theory (DFT) because this theory includes electron correlation in an unspecified way and does not lead to a hierarchy of methods, which lead with increasing sophistication to increasing accuracy of calculated results.
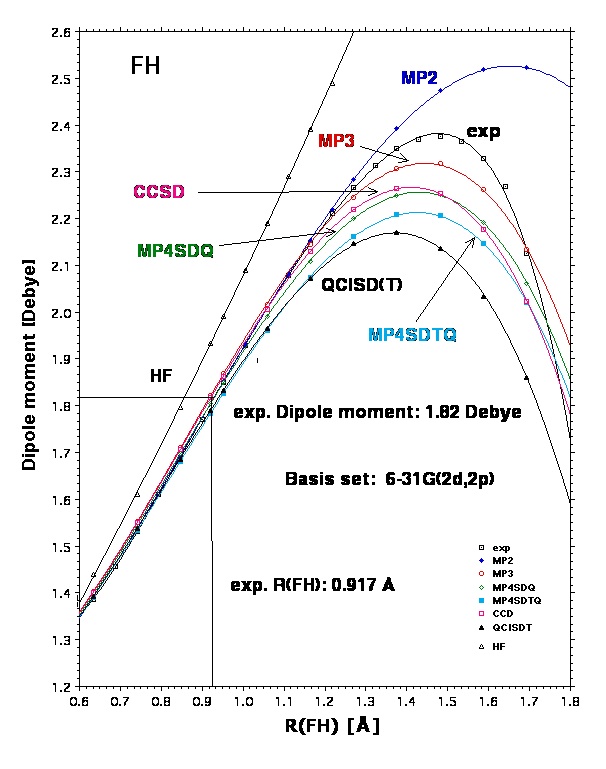
MBPT was used with the Møller-Plesset (MP) perturbation operator at increasing orders n of perturbation theory (MPn series). The MPn series is known to oscillate at lower orders for many molecular properties such as energy, geometry, dipole moment, etc. The even order methods such as second (MP2), fourth (MP4), and sixth order MP (MP6) are the more attractive MP methods because they include always new correlation effects (pair correlation at MP2; orbital relaxation and three-electron correlation at MP4, connected four-electron correlations, disconnected 5- and 6-electron correlations at MP6), which lead to a better quantum chemical description while the odd order MPn methods introduce coupling corrections between correlation effects newly introduced at order n-1. Cremer and He showed that extrapolation of even order MPn methods and that of odd order MPn methods leads for energies to the same infinite order limit identical (or close to) the full CI (FCI) value.
A better description of correlation effects is accomplished by projected CC methods because these contain infinite order correlation effects. For example, the D excitations in CCD cover all possible disconnected pair correlation effects for a given molecule while an extension to single (S) excitations as in CCSD includes also all infinite order orbital relaxation effects. The addition of all infinite order three-electron correlation effects as described by the T excitations is obtained in CCSDT, which is a very accurate but also a very expensive method (cost being proportional to Niter O(M8) where Niter defines the number of iterations needed in order to obtain self-consistent CC amplitudes). Accordingly, one compromises mostly by adding T excitations in a perturbative way to CCSD thus yielding CCSD(T), which scales with Niter O(M7) and, therefore, can be applied for larger M (see above). Since CCSD(T) covers about 70 % of the T effects [Cremer and Z. He] contained in the more complete CCSDT method, it provides reasonable estimates of CCSDT geometries.
On the basis of this investigation we answered a number of interesting questions:
- Does MP6 lead to reliable molecular geometries or do we have to apply even higher orders of MPn theory for this purpose?
- Which correlation effects have to be covered to obtain accurate geometries?
- How do MP and CC methods perform in comparison?
- Is CCSD(T) sufficient to determine accurate geometries?
- What is the most economic way to calculate reliable geometries?
Reference:
- 1J. Gauss, D. Cremer, and J.F. Stanton, J. Chem. Phys. A, 104, 1319 (2000).
- 2 a)Y. He and D. Cremer, J. Phys. Chem. A, 104 (2000) 7679. (b) Y. He and D. Cremer, J. Mol. Phys., 98 (2000) 1415.
- 2 c)Y. He and D. Cremer, Theoret. Chem. Accounts, 105, 101 (2000).
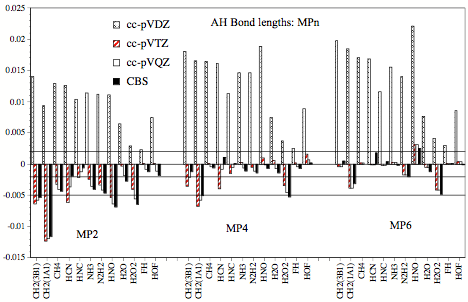
Figure 1. Errors (relative to experimental r_e values) of calculated AH bond lengths obtained at the MP2, MP4, and MP6 level of theory for the cc-pVDZ, cc-pVTZ, cc-pVQZ, and cc-pV∞Z (CBS limit) basis sets.
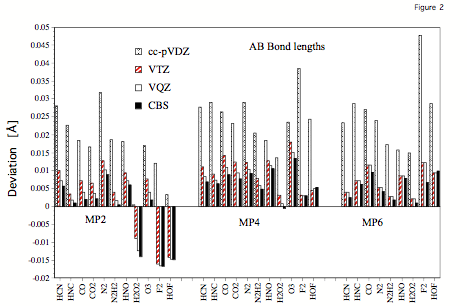
Figure 2. Errors (relative to experimental re values) of calculated AB bond lengths obtained at the MP2, MP4, and MP6 level of theory for the cc-pVDZ, cc-pVTZ,cc-pVQZ, and cc-pV∞Z (CBS limit) basis sets.
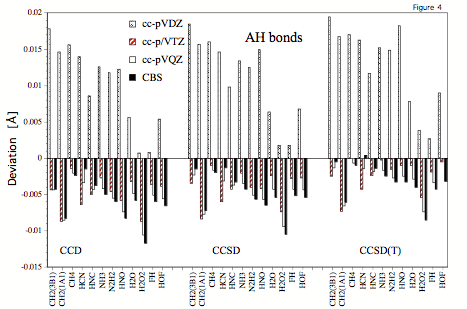
Figure 4. Errors (relative to experimental re values) of calculated AH bond lengths obtained at the CCD, CCSD, and CCSD(T) level of theory for the cc-pVDZ, cc-pVTZ, cc-pVQZ, and cc-pV∞Z (CBS limit) basis sets.
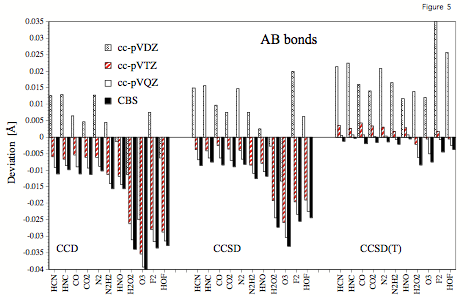
Figure 5. Errors (relative to experimental r_e values) of calculated AB bond lengths obtained at the CCD, CCSD, and CCSD(T) level of theory for the cc-pVDZ, cc-pVTZ, cc-pVQZ, and cc-pV∞Z (CBS limit) basis sets.
XI) Free Silylium Cations
Search for Silyl Cations in Condensed Phases
(Professor Dieter Cremer, Professor Elfi Kraka)
Silicon is more electropositive than carbon and, therefore, silylium cations, SiR3+, are more stable than carbocations in the gas phase. However, in condensed phases trivalent silylium cations are normally not observed whereas carbocations are relatively stable and show a rich chemistry in this environment. The search for silylium cations in condensed phases was controversy discussed and involved as leading figures prominent chemists such as J.B. Lambert, G.A. Olah, and P. v. R. Schleyer. Over years a scientific feud took place between those who believed to have synthesized silylium cations for the first time and those who vividly denied the existence of silylium cations in condensed phases.
Since proof or disproof of the various claims was difficult to obtain by experimental means, quantum chemical investigations provided reliable evidence on the existence of silylium cations in solutions. It turned out that d29Si chemical shift calculations in connection with NMR measurements provided decisive information on the degree of complexation of a silyl cation in solution. Comparison of calculated and measured shift values helped to identify a silyl cation-solvent complex and to describe its structural, energetic, and electronic features on the basis of ab initio calculations.
We could show that in several cases claims of having synthesized free silylium cation in solution were not correct. We worked out the concept of internal solvation and found that even alkanes react with silylium cations. However, we could also demonstrate that trimesitylsilylium cations synthesized by Lambert in benzene solution are indeed free silylium cations in solution.
For further reference, see:
- 133Evidence for the Existence of Silylium Cations in Condensed Phases, L. Olsson and D. Cremer, Chem. Phys. Lett., 215, 433 (1993).
- 146Properties of R3SiX Compounds and R3Si+ Ions: Do Silylium Ions exist in Solution?, L. Olsson, C.-H. Ottosson, and D. Cremer, J. Am. Chem. Soc., 117, 7460 (1995).
- 161Solvated Silylium Cations: Structure Determination by NMR Spectroscopy and the NMR/ab initio/IGLO Method, M. Arshadi, D. Johnels, U. Edlund, C.-H. Ottosson and D. Cremer, J. Am. Chem. Soc. 118, 5120 (1996).
- 163Intramolecularly Stabilized Phenylsilyl and Anthrylsilyl Cations., C.-H. Ottosson and D. Cremer, Organometalics 15, 5309 (1996).
- 164Nature of the Si(SiMe3)3+ Cation in Aromatic Solvents, C.-H. Ottosson and D. Cremer, Organometallics 15, 5495 (1996).
- 180Trimesitylsilylium Cation: Verification of a Free Silylium Cation in Solution by NMR Chemical Shift Calculations, E. Kraka, C.P. Sosa, J. Gräfenstein, and D. Cremer, Chem. Phys. Lett., 279, 9 (1997).
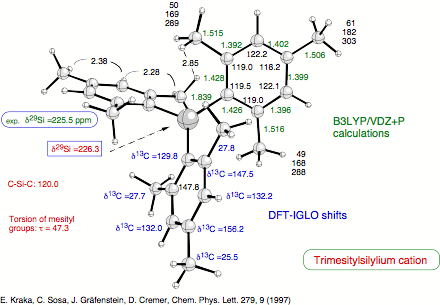
Figure 1. Calculated NMR chemical shifts of the trimesitylsilylium cation. The agreement of the calculated d29Si chemical shift value with the measured one verifies that the calculated geometry represents that of the cation investigated in solution by Lambert. Thereby, the existence of a free silylium cation in solution is verified.
XII) Electron Density Analysis
Description of Chemical Bonding with the help of Electron Density, Energy Density, and Associated Laplace Distributions.
(Professor Elfi Kraka, Professor Dieter Cremer, and co-workers)
Investigation of the electron density distribution, its associated Laplace distribution, and the energy density distribution leads to a unique definition and description of atoms in molecules, chemical bonds, molecular structure, and structural changes. From the local features of electron and energy distribution valuable insight into the bonding mechanism and the reactive behaviour of a molecule is gained, in particular into distortions of the valence shell of an atom upon bond formation; the order, p-character, polarity, and bent character of a bond; the degree of electron delocalization and bond equalization in a conjugated p-system; the extend of through-bond and through-space interactions; the sites that are prone to electrophilic or nucleophilic attack; and the bonds that can easily be broken in a molecule. Important chemical concepts or models such as p- and s-aromaticity, homoaromaticity, ring strain, the frontier orbital model or the HSAB concept can be reevaluated on the basis of the density analysis.
We extended the electron density analysis to nonclassical bonding situations and to describe the beginning of bonding in the transition state. For this purpose, correlation corrected response densities (MBPT and CC) were calculated and analyzed using extended basis sets with s-, p-, d-, and f-functions.
For further reference, see:
- 106Chemical Implication of Local Features of the Electron Density Distribution, E. Kraka and D. Cremer, in "Theoretical Models of Chemical Bonding. The Concept of the Chemical Bond", Z.B. Maksic, Edt., Volume 2, Springer Verlag, Heidelberg, 1990, p.453.
- 111Determination and Use of Response-Densities, E. Kraka, J. Gauss, and D. Cremer, "40 Years in Quantum Chemistry", Special Issue of Journal of Molecular Structure (Theochem), 234, 95 (1991).
- 122Description of Chemical Reactions in Terms of the Properties of the Electron Density, E. Kraka and D. Cremer, "Electron Density and Chemical Reactions", Special Issue of Journal of Molecular Structure (Theochem), 255, 189 (1992).