VI) Development and Application of Relativistic Methods
3. Bonding in Radonhexafluoride: An unusual Relativistic Problem
Radon hexafluoride is a bound species (bond length Rn-F: 2.008 Å) as demonstrated by correlation corrected relativistic ab initio calculations using IORAmm (Infinite Order Regular Approximation with modified metric) at the MP2 level of theory with a (24s20p13d8f) [15s13p8d4f]/aug-cc-pVDZ basis set. The calculated atomization energy is 226.9 kcal/mol and the dissociation energy leading to Rn and 3 F_2 is 126.6 kcal/mol. Results are in line with simple orbital-based predictions of possible relativistic effects. The relativistic effect for the atomization energy is -10.8 kcal/mol rather than +27.7 kcal/mol as predicted on the basis of DHF calculations. The latter were flawed by the lack of correlation corrections and an erroneous polynomial fit of the potential energy surface in the vicinity of the global minimum.
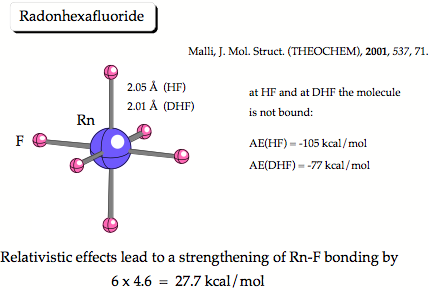
Figure 1. Results of Mali from 2001. He predicts that relativistic effects strengthen the RF bonds. However, this prediction is based on shaky arguments because neither HF nor Dirc-HF theory predicts the molecule to be bound.
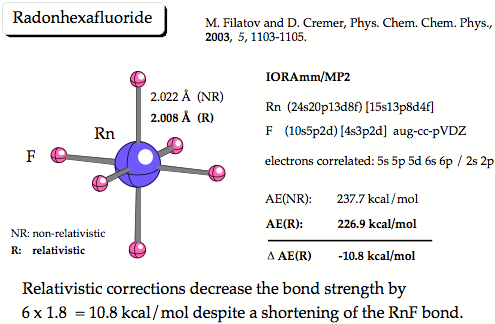
Figure 2. IORAmm/MP2 calculations predict the molecule to be bound by 227 kcal/mol (40 kal/mol per RnF bond). Relativistic effects decrease the bond strength by 1.8 kcal/mol per bond whereas shortening the bond at the same time.
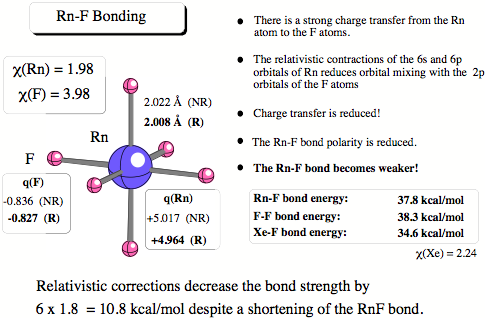
Figure 3. Charge transfer is largely responsible for the strength of the RnF bond. Relativistic effects lead to a contraction of Radon’s 6s and 6p orbitals and in this way charge transfer and the bond strength is reduced whereas the contraction decreases the covalent radius of Rn and by this causes bond shortening.
4. Calculation of Relativistically Corrected Spin-Spin Coupling Constants
(Project leader: Michael Filatov, Dieter Cremer)
Nuclear Magnetic Resonance (NMR) spectroscopy is one of the most important experimental tools in chemistry as is documented in dozens of monographs and review articles. During the last ten years, quantum chemistry has taken this into account by focusing on the development of methods for calculating NMR parameters such as chemical shieldings or spin-spin coupling constants (SSCCs). The reliable prediction of SSCCs on a routine basis represented for a long time a major obstacle, however this has been largely solved in the last years. Coupled-perturbed (CP) DFT predictions of SSCCs have proven in many cases to be reliable and, because of its favorable cost/efficiency ratio, CP-DFT is the method of choice for calculating SSCCs of larger molecules.
One of the remaining problems is the satisfactory calculation of SSCC involving heavy nuclei. In such a case, there is a need for relativistic corrections especially of the FC term to obtain a reasonable description of spin polarization at the contact surface of the nuclei.
Recently, we have developed a new computational procedure for the calculation of the Hamiltonian matrix elements within the regular approximation for relativistic effects. The new procedure is fully analytic and thus it can be applied in the context of both WFT and DFT calculations. With the use of the new procedure, quasi-relativistic computational schemes more accurate than ZORA, such as infinite-order regular approximation (IORA) and infinite-order regular approximation with modified metric (IORAmm), can be installed easily within the standard quantum-chemical codes designed for non-relativistic calculations.
Based on these developments we worked out a computationally inexpensive quasi-relativistic method for calculating nuclear SSCCs in molecules containing heavy elements. In this way, our IORAmm formalism for the calculation of molecular properties was extended to the calculation of SSCCs as new second-order response property. Most successful in this connection was the use of CP-DFT and the corresponding method was coined IORAmm/CP-DFT/SSCC. Since IORAmm/CP-DFT/SSCC is as fast as the corresponding non-relativistic CP-DFT/SSCC method , new possibilities of investigating relatively large molecular systems with heavy atoms are opened up. In this project, we will calculate SSCC for group IVa compounds involving Ge, Sn, and Pb and in this way to complement SSCC, which are experimentally known by calculated ones. Moreover, we will focus on molybdenum and tungsten compounds for which experimental SSCCs have been used to explain bonding. In both cases, we will combine experimental and calculated information to identify structural and bonding features questionable so far on the available experimental data. In this connection, we will also carry out a basic studies answering the question, which functional is best suited for the relativistic SSCC calculations. Since GGA functionals have a singularity at the nucleus, we will seek for ways of eliminating errors arising from these singularities.